
profesional sanitario
El perfil de seguridad de KOSELUGO® en monoterapia se ha determinado tras evaluar una población combinada de 74 pacientes pediátricos con Neurofibromatosis Tipo 1 (NF1) que presentan neurofibromas plexiformes (NPs) sintomáticos e inoperables - 24 pacientes de la fase I del ensayo SPRINT (N = 24; 20-30 mg/m2 de superficie corporal (SC) dos veces al día) y 50 pacientes del estrato 1 de la fase II del ensayo SPRINT (N = 50; 25 mg/m2 de SC dos veces al día).1-3
El 61% (N = 45/74) de los pacientes fueron tratados durante > 48 meses.1 Para la monitorización de la seguridad y la eficacia a largo plazo, los pacientes del estrato 1 de la fase II de SPRINT permanecieron en el estudio 7 años des del inicio del tratamiento con KOSELUGO® o 5 años des de la interrupción del tratamiento con KOSELUGO®, lo que fuera más largo.2
La mediana de la duración total del tratamiento con KOSELUGO® en pacientes pediátricos con NF1-NP fue de 55 meses (rango: < 1 a 97 meses).1-3
No hubo diferencias clínicamente relevantes en el perfil de seguridad entre el estudio fase I SPRINT y el estrato 1 de la fase II de SPRINT.1-3
Tabla de: Ficha Técnica de KOSELUGO®.1
*Identificado a partir de la experiencia de otros ensayos clínicos en pacientes adultos (N = 347) con múltiples tipos de tumores, que recibieron KOSELUGO® (75 mg dos veces al día). Estos EAs no se han informado en pacientes pediátricos con NF1 y NP inoperable.1
EA: efecto adverso; ALT: alanina aminotransferasa; AST: aspartato aminotransferasa; CPK: creatina fosfoquinasa; RSC: retinopatía serosa central; DEPR: desprendimiento del epitelio pigmentario de la retina; OVR: oclusión venosa retiniana; SOC: Clasificación por grupos y sistemas; MedDRA: diccionario médico para actividades reglamentarias; CTCAE: Criterios terminológicos comunes para acontecimientos adversos.
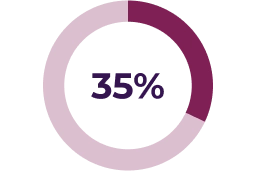
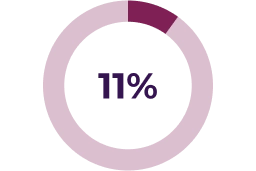
Se han notificado descensos asintomáticos de la fracción de eyección en el 26 % de los pacientes pediátricos del ensayo clínico pivotal. La mediana del tiempo hasta la aparición inicial de estas reacciones adversas fue de 232 días. Se ha reportado algunos informes graves de reducción de la FEVI asociada con KOSELUGO® en pacientes pediátricos que participaron en un programa de acceso expandido.
No se han estudiado pacientes pediátricos con antecedentes de deterioro de la función ventricular izquierda o una FEVI basal por debajo del nivel institucional inferior normal (LIN). La FEVI debe evaluarse mediante ecocardiograma antes de iniciar el tratamiento para establecer valores basales. Antes de iniciar el tratamiento con KOSELUGO®, los pacientes deben tener una fracción de eyección superior al LIN institucional.
La FEVI se debe evaluar a intervalos de aproximadamente 3 meses, o con mayor frecuencia si está clínicamente indicado durante el tratamiento. La reducción de la FEVI se puede controlar mediante la interrupción del tratamiento, la reducción de la dosis o la suspensión permanente del tratamiento.
Se debe aconsejar a los pacientes que notifiquen cualquier alteración visual nueva. Se han notificado reacciones adversas de visión borrosa en pacientes pediátricos que recibieron KOSELUGO®. Se han observado casos aislados de desprendimiento del epitelio pigmentario de la retina (DEPR), retinopatía serosa central (RSC) y oclusión venosa retiniana (OVR) en pacientes adultos con varios tipos de tumores que recibieron tratamiento con KOSELUGO® en monoterapia y en combinación con otros antineoplásicos, y en un único paciente pediátrico con astrocitoma pilocítico tratado con KOSELUGO® en monoterapia.
De acuerdo con la práctica clínica, se recomienda una exploración oftalmológica antes de comenzar el tratamiento y en cualquier momento en que un paciente refiera nuevos trastornos visuales. A los pacientes a los que se les ha diagnosticado DEPR o RSC sin reducción de la agudeza visual, se les debe realizar una exploración oftalmológica cada 3 semanas hasta la resolución. Si se diagnostica DEPR o RSC y la agudeza visual está afectada, se deberá interrumpir el tratamiento con KOSELUGO® y reducir la dosis al reanudar el tratamiento. Si se diagnostica OVR, el tratamiento con KOSELUGO® se deberá suspender de forma permanente.
Los estudios de interacciones se han realizado solo en adultos sanos (edad ≥ 18 años).
Principios activos que pueden aumentar las concentraciones plasmáticas de KOSELUGO®La administración conjunta con un inhibidor potente de la CYP3A4 (200 mg de itraconazol dos veces al día durante 4 días) aumentó la concentración plasmática máxima (Cmax) de KOSELUGO® en un 19% (IC 90%: 4, 35) y el área bajo la curva (AUC) en un 49% (IC 90%: 40, 59) en adultos sanos.
La administración conjunta con un inhibidor potente de la CYP2C19 o un inhibidor moderado de la CYP3A4 (200 mg de fluconazol una vez al día durante 4 días) aumentó la Cmax de KOSELUGO® en un 26% (IC 90%: 10, 43) y el AUC en un 53% (IC 90%: 44, 63) en adultos sanos, respectivamente.
Se prevé que el uso concomitante de eritromicina (inhibidor moderado de la CYP3A4) o fluoxetina (inhibidor potente de la CYP2C19/CYP2D6) aumentará el AUC de KOSELUGO® en un ~30-40% y la Cmax en un ~20%.
Se debe evitar la administración conjunta con inhibidores potentes de la CYP3A4 (p. ej., claritromicina, zumo de pomelo, ketoconazol oral) o la CYP2C19 (p. ej., ticlopidina). Se debe evitar la administración conjunta con inhibidores moderados de la CYP3A4 (p. ej., eritromicina y fluconazol) y de la CYP2C19 (p. ej., omeprazol).
No se recomienda la administración conjunta de inhibidores potentes o moderados de la CYP3A4 o CYP2C19 y se deberían considerar agentes alternativos.
Si la administración conjunta es inevitable, se debe hacer un seguimiento riguroso de los pacientes por si presentan acontecimientos adversos y se debe reducir la dosis de KOSELUGO®:Principios activos que pueden reducir las concentraciones plasmáticas de KOSELUGO®
La administración conjunta con un inductor potente de la CYP3A4 (600 mg diarios de rifampicina durante 8 días) disminuyó la Cmax de KOSELUGO® en un −26 % (IC 90%: −17, −34) y el AUC en un −51% (IC 90%: −47, −54).
Se debe evitar el uso concomitante de inductores potentes de la CYP3A4 (p. ej., fenitoína, rifampicina, carbamazepina, hipérico) o inductores moderados de la CYP3A4 con KOSELUGO®.
Principios activos cuyas concentraciones plasmáticas pueden ser alteradas por KOSELUGO®In vitro, KOSELUGO® es un inhibidor del OAT3. No se puede descartar la posibilidad de un efecto clínicamente relevante sobre la farmacocinética de los sustratos de OAT3 administrados de forma concomitante (p. ej., metotrexato y furosemida).
El TPGS es un inhibidor de la P-gp in vitro y no se puede excluir que pueda causar interacciones medicamentosas clínicamente relevantes con sustratos de la P-gp (p. ej., digoxina o fexofenadina).
No se ha evaluado el efecto de KOSELUGO® sobre la exposición de anticonceptivos orales. Por tanto, se debe recomendar el uso de un método de barrera adicional a las mujeres que utilizan anticonceptivos hormonales.
Efecto de los antisecretores gástricos sobre KOSELUGO®Las cápsulas de KOSELUGO® no presentan una disolución dependiente del pH. KOSELUGO® se puede usar concomitantemente con fármacos modificadores del pH gástrico (p. ej., antagonistas del receptor H2 e inhibidores de la bomba de protones) sin restricciones, excepto con omeprazol, que es un inhibidor de la CYP2C19.
▼ Este medicamento está sujeto a seguimiento adicional, es prioritaria la notificación de sospecha de reacciones adversas asociadas a este medicamento.
PRESENTACIÓN Y PRECIO: Koselugo® 10 mg cápsulas duras, 60 cápsulas. C.N: 731322. PVL notificado: 5.400 €. Koselugo® 25 mg cápsulas duras, 60 cápsulas. C.N: 731324. PVL notificado: 13.500 €. Financiación por el S.N.S restringida a determinadas indicaciones/condiciones.
RÉGIMEN DE PRESCRIPCIÓN Y DISPENSACIÓN: Medicamento sujeto a prescripción médica. Diagnóstico hospitalario.
Consulte la Ficha Técnica completa antes de prescribir este medicamento.