
La deficiencia de lipasa ácida lisosomal (LAL-D) es una enfermedad rara, de naturaleza progresiva y potencialmente mortal.1,2
Su herencia es autosómica recesiva y es causada por mutaciones en el gen LIPA que interrumpen la actividad de la enzima lipasa ácida lisosomal (LAL).1-3
La edad de inicio es muy variable, pudiéndose presentar en lactantes, niños y adultos con complicaciones metabólicas que pueden ser graves.1,3
Los lactantes presentan la progresión más rápida que puede resultar en muerte generalmente dentro de los primeros 6-12 meses de vida.1,3
Su herencia es autosómica recesiva y es causada por mutaciones en el gen LIPA que interrumpen la actividad de la enzima lipasa ácida lisosomal (LAL).1-3
La edad de inicio es muy variable, pudiéndose presentar en lactantes, niños y adultos con complicaciones metabólicas que pueden ser graves.1,3
Los lactantes presentan la progresión más rápida que puede resultar en muerte generalmente dentro de los primeros 6-12 meses de vida.1,3
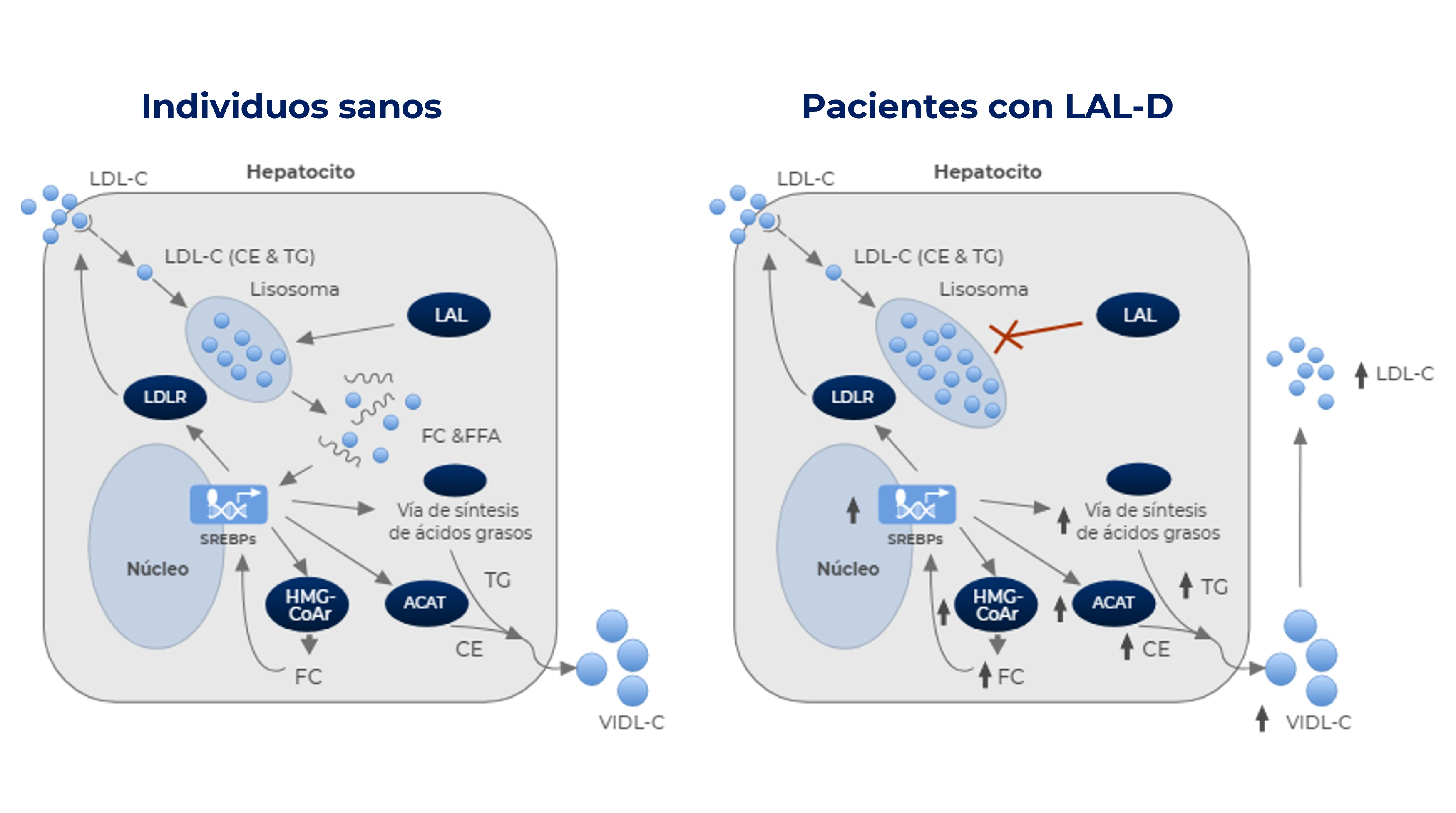
La enzima LAL es esencial para la hidrólisis de los ésteres de colesterol y triglicéridos en los lisosomas.1,4
Como consecuencia de la LAL-D, los lípidos se acumulan progresivamente en el interior de los lisosomas, lo que conduce a la disfunción celular en múltiples órganos y tejidos vitales, como el hígado o el bazo, entre otros órganos.1-2,4
Existen diferencias a nivel de homeostasis del colesterol celular en individuos sanos y en pacientes con LAL-D:2
Como consecuencia de la LAL-D, los lípidos se acumulan progresivamente en el interior de los lisosomas, lo que conduce a la disfunción celular en múltiples órganos y tejidos vitales, como el hígado o el bazo, entre otros órganos.1-2,4
Existen diferencias a nivel de homeostasis del colesterol celular en individuos sanos y en pacientes con LAL-D:2
{kind=link}
LAL-D es una enfermedad heterogénea, con signos y síntomas de progresión muy variable entre los individuos afectados.2,4




Marcadores séricos4


*Relacionados con LAL-D rápidamente progresiva.
ALT: alanina aminotransferasa; AST: aspartato transaminasa; HDL: lipoprotína de alta densidad; LAL: lipasa ácida lisosomal; LAL-D: deficiencia de lipasa ácida lisosomal; LDL: lipoproteína de baja densidad; LIPA: lipasa A, tipo ácido lisosomal; TG: triglicéridos.
profesional sanitario
Conoce más sobre las asociaciones de pacientes en España relacionadas con esta patología.
AELALD está constituida por pacientes,
familiares y amigos de pacientes afectados por
LAL-D. Su creación es fruto de la necesidad de
organizarse y sumar esfuerzos dado el bajo
número de casos diagnosticados en base a las
posibles prevalencias.
familiares y amigos de pacientes afectados por
LAL-D. Su creación es fruto de la necesidad de
organizarse y sumar esfuerzos dado el bajo
número de casos diagnosticados en base a las
posibles prevalencias.