
Consulte la Ficha Técnica de ULTOMIRIS® antes de prescribir este medicamento.



¶La respuesta completa de la MAT se definió como la normalización del recuento de plaquetas (≥ 150 × 109/l), la normalización de la LDH sérica (≤ 246 U/l) y una mejora ≥ 25 % en la creatinina sérica respecto al valor inicial.1
#Entre los pacientes con datos evaluables, ULTOMIRIS® permitió la recuperación de la función renal (mejora en ≥ 1 estadio de la categoría de TFGe) frente al valor inicial, en el 70 % (n = 30/43) de los pacientes adultos y en el 100 % (n = 16/16) de los pacientes pediátricos y adolescentes después de 1 año. 17 de los 29 pacientes que requirieron diálisis al ingresar al estudio, pudieron suspender la diálisis al final del seguimiento disponible, mientras que 6 de los 27 pacientes que no estaban en diálisis al inicio, estaban en diálisis al final del período de evaluación inicial.1 Entre los pacientes con SHUa que, al inicio, estaban en terapia de remplazo renal (TRR), el tratamiento con ULTOMIRIS® condujo a la interrupción de la diálisis en el 59 % de los pacientes adultos (n = 17/29; seguimiento medio de 76,7 semanas) y en el 100 % de los pacientes pediátricos (n = 6/6; seguimiento de 50 semanas).3,5
SHUa: síndrome hemolítico urémico atípico; C5: proteína 5 del complemento; MAT: microangiopatía trombótica; LDH: lactato deshidrogenasa; TRR: terapia de remplazo renal.
Los resultados de los intercambios plasmáticos (PE) o infusiones de plasma (PI) que reciben pacientes con SHUa son pobres; entre el 45 % y el 56 % de los pacientes adultos progresan a enfermedad renal crónica terminal (ERCT) o mueren en el plazo de 1 año,9,10 y hasta el 77 % tienen ERCT o mueren en el plazo de 3 años.11
La identificación rápida de la MAT, el diagnóstico del SHUa y el inicio urgente de un tratamiento específico son cruciales para preservar la función renal y mejorar los resultados.6,12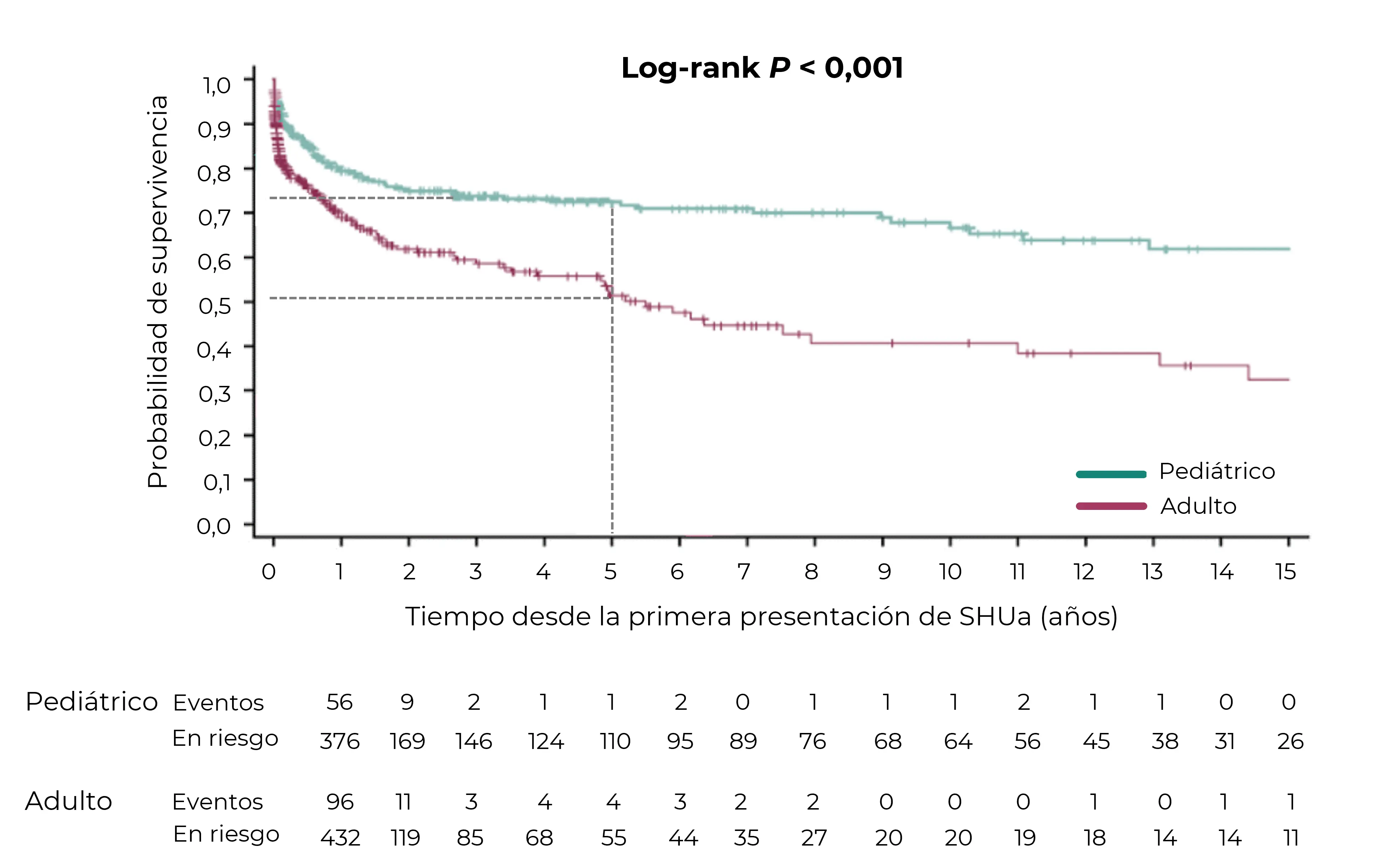{kind=link}
Estimaciones acumuladas de Kaplan-Meier para la probabilidad de supervivencia de la ERCT analizadas según la edad adulta y pediátrica en la presentación inicial.
Número de pacientes en riesgo mostrado cada año después de la presentación inicial de SHUa.
El análisis de la ERCT excluye a aquellos pacientes con trasplante de riñón antes de la presentación de SHUa (n = 36) y aquellos con inicio de eculizumab en la presentación de SHUa (n = 7).13
SHUa: síndrome hemolítico urémico atípico; MAT: microangiopatía trombótica; PE: intercambio plasmático; PI: infusión de plasma; ERCT: enfermedad renal crónica terminal.
46 % (n = 57/125)
71 % (n = 89/125)
17 % (n = 15/89)
39 % (n = 35/89)
†Seguimiento medio de 57 meses (rango, 1-353 meses).9
‡Seguimiento medio de 45 meses (rango, 1-493 meses).9
SHUa: síndrome hemolítico urémico atípico; MAT: microangiopatía trombótica; ERCT: enfermedad renal crónica terminal.

PRESENTACIÓN Y PRECIO. Ultomiris 300 mg/3 ml concentrado para solución para perfusión, 1 vial de 3 ml. C.N: 731120. PVL notificado: 5.018 €. Ultomiris 1100 mg/11 ml concentrado para solución para perfusión, 1 vial de 11 ml. C.N: 731121. PVL notificado: 18.399,33 €. Financiación por el S.N.S para determinadas indicaciones/condiciones. La indicación de Trastorno del espectro de neuromielitis óptica (TENMO) no está incluida en la prestación farmacéutica del Sistema Nacional de Salud. RÉGIMEN DE PRESCRIPCIÓN Y DISPENSACIÓN. Medicamento sujeto a prescripción médica. Uso Hospitalario. Consulte la Ficha Técnica completa antes de prescribir este medicamento.
profesional sanitario