
healthcare professional
KOSELUGO® is indicated for the treatment of symptomatic, inoperable PN in paediatric patients with NF1 aged ≥ 3 years.1 Please refer to the KOSELUGO® SmPC for full prescribing information
The safety of KOSELUGO® was evaluated in a combined population of 74 paediatric patients with neurofibromatosis type 1 with inoperable, symptomatic plexiform neurofibroma (NF1-PN) – from the SPRINT Phase I study (N = 24; 20–30 mg/m2 body surface area [BSA] twice daily) and the SPRINT Phase II Stratum 1 study (N = 50; 25 mg/m2 BSA twice daily).1
61% (n = 45/74) of patients were treated for > 48 months.1 Patients in SPRINT Phase II Stratum 1 remained on study for long-term safety and efficacy monitoring for 7 years following initiation of KOSELUGO® treatment or 5 years after KOSELUGO® discontinuation, whichever was longer.2
The median total duration of KOSELUGO® treatment in paediatric patients with NF1-PN was 55 months (range: < 1–97 months).1
There were no clinically relevant differences in the safety profile between the SPRINT Phase I and Phase II Stratum 1 studies.1
61% (n = 45/74) of patients were treated for > 48 months.1 Patients in SPRINT Phase II Stratum 1 remained on study for long-term safety and efficacy monitoring for 7 years following initiation of KOSELUGO® treatment or 5 years after KOSELUGO® discontinuation, whichever was longer.2
The median total duration of KOSELUGO® treatment in paediatric patients with NF1-PN was 55 months (range: < 1–97 months).1
There were no clinically relevant differences in the safety profile between the SPRINT Phase I and Phase II Stratum 1 studies.1

The most commonly reported AEs were Grade 1 and 2 gastrointestinal symptoms (vomiting, nausea or diarrhoea), an asymptomatic increase in creatine phosphokinase levels, acneiform rash and paronychia.3
AE, adverse event; AST, aspartate aminotransferase; CPK, creatine phosphokinase.
Table adapted from KOSELUGO® SmPC 2024.1
*Identified from other clinical trial experience in adult patients (N = 347) with multiple tumour types, receiving KOSELUGO® (75 mg twice daily). These AEs have not been reported in paediatric patients with NF1 and inoperable PN.1
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; CSR, central serous retinopathy; RPED, retinal pigment epithelial detachment; RVO retinal vein occlusion.
*Identified from other clinical trial experience in adult patients (N = 347) with multiple tumour types, receiving KOSELUGO® (75 mg twice daily). These AEs have not been reported in paediatric patients with NF1 and inoperable PN.1
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; CPK, creatine phosphokinase; CSR, central serous retinopathy; RPED, retinal pigment epithelial detachment; RVO retinal vein occlusion.
In SPRINT Phase I and Phase II Stratum 1:2
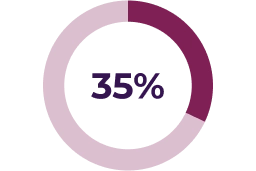
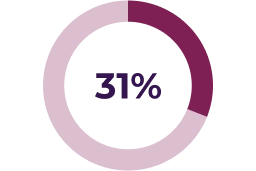
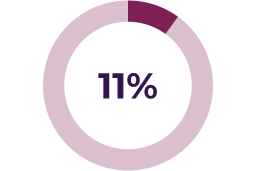
Asymptomatic decreases in ejection fraction have been reported in 26% of paediatric patients in the pivotal clinical trial. Median time to initial onset of these adverse reactions was 232 days. A small number of serious reports of LVEF reduction associated with KOSELUGO® have been reported in paediatric patients who participated in an expanded access program.
Paediatric patients with a history of impaired left ventricular function or a baseline LVEF below institutional lower level of normal (LLN) have not been studied. LVEF should be evaluated by echocardiogram before initiation of treatment to establish baseline values. Before starting KOSELUGO® treatment, patients should have an ejection fraction above the institutional LLN. LVEF should be evaluated at approximately 3-month intervals, or more frequently as clinically indicated, during treatment. Reduction in LVEF can be managed using treatment interruption, dose reduction or treatment discontinuation.
Paediatric patients with a history of impaired left ventricular function or a baseline LVEF below institutional lower level of normal (LLN) have not been studied. LVEF should be evaluated by echocardiogram before initiation of treatment to establish baseline values. Before starting KOSELUGO® treatment, patients should have an ejection fraction above the institutional LLN. LVEF should be evaluated at approximately 3-month intervals, or more frequently as clinically indicated, during treatment. Reduction in LVEF can be managed using treatment interruption, dose reduction or treatment discontinuation.
Patients should be advised to report any new visual disturbances. Adverse reactions of blurred vision have been reported in paediatric patients receiving KOSELUGO®. In adult patients with multiple tumour types, receiving treatment with KOSELUGO® monotherapy, and in combination with other anti-cancer agents, and in a single paediatric patient with pilocytic astrocytoma on KOSELUGO® monotherapy, have been observed.
In line with clinical practice, an ophthalmological evaluation before treatment initiation and at any time a patient reports new visual disturbances is recommended. In patients diagnosed with retinal pigment epithelial detachment (RPED) or central serous retinopathy (CSR) without reduced visual acuity, ophthalmic assessment should be conducted every 3 weeks until resolution. If RPED or CSR is diagnosed and visual acuity is affected, KOSELUGO® therapy should be interrupted, and the dose reduced when treatment is resumed. If retinal vein occlusion (RVO) is diagnosed, treatment with KOSELUGO® should be permanently discontinued.
In line with clinical practice, an ophthalmological evaluation before treatment initiation and at any time a patient reports new visual disturbances is recommended. In patients diagnosed with retinal pigment epithelial detachment (RPED) or central serous retinopathy (CSR) without reduced visual acuity, ophthalmic assessment should be conducted every 3 weeks until resolution. If RPED or CSR is diagnosed and visual acuity is affected, KOSELUGO® therapy should be interrupted, and the dose reduced when treatment is resumed. If retinal vein occlusion (RVO) is diagnosed, treatment with KOSELUGO® should be permanently discontinued.
Liver laboratory abnormalities, specifically aspartate aminotransferase and alanine aminotransferase elevations, can occur with KOSELUGO®. Liver laboratory values should be monitored before initiation of KOSELUGO® and at least monthly during the first 6 months of treatment, and thereafter as clinically indicated. Liver laboratory abnormalities should be managed with dose interruption or reduction or treatment discontinuation.
Skin rash (including maculopapular rash and acneiform rash), paronychia and hair changes have been reported very commonly in the pivotal clinical study. Dry skin, hair colour changes, paronychia and rash maculo-papular were seen more frequently in younger children (age 3–11 years) and acneiform rash was seen more frequently in post-pubertal children (age 12–16 years).
KOSELUGO® capsules contain vitamin E. Therefore, patients should be advised not to take any supplemental vitamin E. High doses of vitamin E may increase the risk of bleeding in patients taking concomitant anticoagulant or antiplatelet medicinal products (e.g. warfarin or aspirin). Anticoagulant assessments should be conducted more frequently to detect when dose adjustments of the anticoagulant or antiplatelet medicinal products are warranted.
KOSELUGO® is available as a capsule which must be swallowed whole. Some patients, in particular children <6 years of age, may be at risk of choking on a capsule formulation owing to developmental, anatomical or psychological reasons. Therefore, KOSELUGO® should not be administered to patients who are unable or unwilling to swallow the capsule whole.
KOSELUGO® is not recommended in women of childbearing potential who are not using contraception.
KOSELUGO® is contraindicated in those with hypersensitivity to the active substance or to any of the excipients and those with severe hepatic impairment.
Interaction studies have only been performed in healthy adults (aged ≥ 18 years).
Active substances that may increase KOSELUGO® plasma concentrations
Co-administration with a strong cytochrome P450 (CYP) 3A4 inhibitor (200 mg itraconazole twice daily for 4 days) increased KOSELUGO® maximum plasma concentration (Cmax) by 19% (90% CI: 4, 35) and area under the curve (AUC) by 49% (90% CI: 40, 59) in healthy adults.
Co-administration with a strong CYP2C19/moderate CYP3A4 inhibitor (200 mg fluconazole once daily for 4 days) increased KOSELUGO® Cmax by 26% (90% CI: 10, 43) and AUC by 53% (90% CI: 44, 63) in healthy adults.
Concomitant use of erythromycin (moderate CYP3A4 inhibitor) or fluoxetine (strong CYP2C19/CYP2D6 inhibitor) is predicted to increase KOSELUGO® AUC by ~ 30–40% and Cmax by ~ 20%.
Co-administration with strong inhibitors of CYP3A4 (e.g. clarithromycin, grapefruit juice, oral ketoconazole) or CYP2C19 (e.g. ticlopidine) should be avoided. Co-administration with moderate inhibitors of CYP3A4 (e.g. erythromycin and fluconazole) and CYP2C19 (e.g. omeprazole) should be avoided.
Concomitant use of strong or moderate CYP3A4 or CYP2C19 inhibitors is not recommended and alternative agents should be considered.
If co-administration with a strong or moderate CYP3A4 or CYP2C19 inhibitor is unavoidable, patients should be carefully monitored for adverse events and the KOSELUGO® dose should be reduced as follows:
Co-administration with a strong CYP3A4 inducer (600 mg rifampicin daily for 8 days) decreased KOSELUGO® Cmax by −26% (90% CI: −17, −34) and AUC by −51% (90% CI: −47, −54).
Concomitant use of strong CYP3A4 inducers (e.g. phenytoin, rifampicin, carbamazepine, St. John's Wort) or moderate CYP3A4 inducers with KOSELUGO® should be avoided.
Active substances whose plasma concentrations may be altered by KOSELUGO®
In vitro, KOSELUGO® is an inhibitor of OAT3. The potential for a clinically relevant effect on the pharmacokinetics of concomitantly administered substrates of OAT3 (e.g. methotrexate and furosemide) cannot be excluded.
D-α-tocopheryl polyethylene glycol succinate (TPGS) is a P-gp inhibitor in vitro and it cannot be excluded that it may cause clinically relevant drug interactions with substrates of P-gp (e.g. digoxin or fexofenadine).
The effect of KOSELUGO® on the exposure of oral contraceptives has not been evaluated. Therefore, use of an additional barrier method should be recommended to women using hormonal contraceptives.
Effect of gastric acid reducing agents on KOSELUGO®
KOSELUGO® capsules do not exhibit pH-dependent dissolution. KOSELUGO® can be used concomitantly with gastric pH-modifying agents (i.e. H2-receptor antagonists and proton pump inhibitors) without restrictions, except for omeprazole, which is a CYP2C19 inhibitor.
Active substances that may increase KOSELUGO® plasma concentrations
Co-administration with a strong cytochrome P450 (CYP) 3A4 inhibitor (200 mg itraconazole twice daily for 4 days) increased KOSELUGO® maximum plasma concentration (Cmax) by 19% (90% CI: 4, 35) and area under the curve (AUC) by 49% (90% CI: 40, 59) in healthy adults.
Co-administration with a strong CYP2C19/moderate CYP3A4 inhibitor (200 mg fluconazole once daily for 4 days) increased KOSELUGO® Cmax by 26% (90% CI: 10, 43) and AUC by 53% (90% CI: 44, 63) in healthy adults.
Concomitant use of erythromycin (moderate CYP3A4 inhibitor) or fluoxetine (strong CYP2C19/CYP2D6 inhibitor) is predicted to increase KOSELUGO® AUC by ~ 30–40% and Cmax by ~ 20%.
Co-administration with strong inhibitors of CYP3A4 (e.g. clarithromycin, grapefruit juice, oral ketoconazole) or CYP2C19 (e.g. ticlopidine) should be avoided. Co-administration with moderate inhibitors of CYP3A4 (e.g. erythromycin and fluconazole) and CYP2C19 (e.g. omeprazole) should be avoided.
Concomitant use of strong or moderate CYP3A4 or CYP2C19 inhibitors is not recommended and alternative agents should be considered.
If co-administration with a strong or moderate CYP3A4 or CYP2C19 inhibitor is unavoidable, patients should be carefully monitored for adverse events and the KOSELUGO® dose should be reduced as follows:
- If a patient is currently taking 25 mg/m2 BSA twice daily, reduce dose to 20 mg/m2 BSA twice daily
- If a patient is currently taking 20 mg/m2 twice daily, reduce dose to 15 mg/m^2 BSA twice daily
Co-administration with a strong CYP3A4 inducer (600 mg rifampicin daily for 8 days) decreased KOSELUGO® Cmax by −26% (90% CI: −17, −34) and AUC by −51% (90% CI: −47, −54).
Concomitant use of strong CYP3A4 inducers (e.g. phenytoin, rifampicin, carbamazepine, St. John's Wort) or moderate CYP3A4 inducers with KOSELUGO® should be avoided.
Active substances whose plasma concentrations may be altered by KOSELUGO®
In vitro, KOSELUGO® is an inhibitor of OAT3. The potential for a clinically relevant effect on the pharmacokinetics of concomitantly administered substrates of OAT3 (e.g. methotrexate and furosemide) cannot be excluded.
D-α-tocopheryl polyethylene glycol succinate (TPGS) is a P-gp inhibitor in vitro and it cannot be excluded that it may cause clinically relevant drug interactions with substrates of P-gp (e.g. digoxin or fexofenadine).
The effect of KOSELUGO® on the exposure of oral contraceptives has not been evaluated. Therefore, use of an additional barrier method should be recommended to women using hormonal contraceptives.
Effect of gastric acid reducing agents on KOSELUGO®
KOSELUGO® capsules do not exhibit pH-dependent dissolution. KOSELUGO® can be used concomitantly with gastric pH-modifying agents (i.e. H2-receptor antagonists and proton pump inhibitors) without restrictions, except for omeprazole, which is a CYP2C19 inhibitor.
AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase; AUC, area under the concentration-time curve; CI, confidence interval; CPK, creatine phosphokinase; CSR, central serous retinopathy; CTCAE, Common Terminology Criteria for Adverse Events; LVEF, left ventricular ejection fraction, LLN, lower limit of normal; NF1, type 1 neurofibromatosis; PN, plexiform neurofibromas; RPED, retinal pigment epithelial detachment; RVO, retinal vein occlusion; TPGS, D-α-tocopheryl polyethylene glycol succinate.
▼ This medicinal product is subject to additional monitoring. This will allow quick identification of new safety information.
Please report any adverse events via your national reporting system. Adverse events should also be reported to AstraZeneca by visiting https://contactazmedical.astrazeneca.com or by calling 0800 783 0033.