
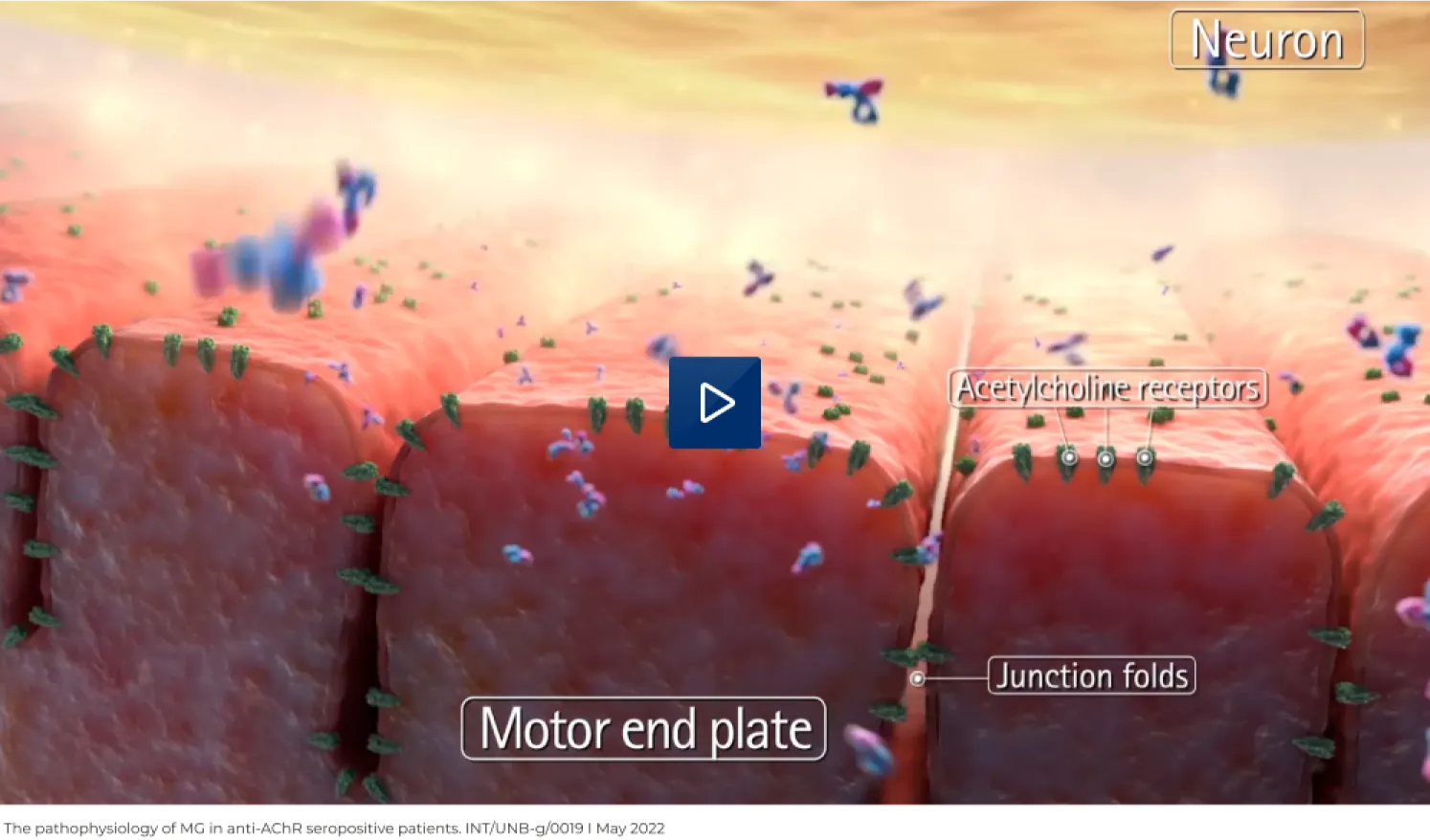
The hallmark clinical feature of myasthenia gravis (MG) is fatigable, fluctuating muscle weakness that declines with repetitive use and improves with rest.2,3 This is caused by the impairment of neuromuscular transmission as a result of the activity of autoantibodies against key molecules functioning at the NMJ.4–6
The binding of autoantibodies to AChRs at the postsynaptic membrane is thought to reduce the functionality of the receptors via three key pathophysiological mechanisms that ultimately reduce their capacity to generate action potentials, causing muscle weakness:7
1. Complement-mediated NMJ destruction
2. Functional AChR blockade
3. Cross-linking of AChRs resulting in their internalisation and destruction (so-called antigenic modulation)
- The binding of anti-AChR antibodies to the AChR triggers the activation of the complement system7
- Terminal complement activation results in the cleavage of C5 into C5a and C5b8
- C5a leads to inflammation, while C5b binds to other complement proteins to form the membrane attack complex (MAC)8
- MAC formation results in destruction of the NMJ7,8
Unlike anti-AChR antibodies, antibodies against the muscle-specific kinase (MuSK) are unable to activate complement directly, but are thought to reduce the numbers of AChR clusters at the postsynaptic membrane by preventing the interaction of MuSK with the low-density lipoprotein receptor-related protein 4 (LRP4).6,9
While the underlying cause for the production of autoantibodies in MG is not fully understood, it is widely agreed that the thymus plays a pivotal role in production of AChR autoantibodies.4,6 Since the thymus acts as a hub for autoimmune activity, it is possible that thymic impairment encourages self-reactive T cell-mediated activation of B cells in patients with anti-AChR antibody positive MG.4,6 Thymic abnormalities, especially hyperplasia and thymoma, are frequently observed in patients with MG, and thymectomy has been demonstrated to enhance outcomes in these patients.5,6 There is likely no involvement of the thymus in anti-MuSK antibody positive MG, and as such, thymectomy does not seem to be beneficial in patients with MuSK MG.4,5
healthcare professional