





{kind=link}
- Reducing leukocyte distribution and trafficking
- Inhibiting lymphocyte recruitment and migration to an inflammatory site
- Blocking T cell function
- Reducing the production and secretion of cytokines and other immune mediators from macrophages or T cells
- Decreasing dendritic cell maturation
Long-term use of corticosteroids is accompanied by several adverse effects.1,10 These include osteoporosis, weight gain, impaired glucose tolerance, skin atrophy, mood disorders, glaucoma and increased risk of infection.1 The number and severity of adverse effects increase with the duration and cumulative dosage of the corticosteroid treatment.8
PE acts by removing key immune molecules from a patient’s plasma, including antibodies, complement components, cytokines and various adhesion molecules.1 IA is a more selective technique than PE, as it removes circulating immunoglobulin G (IgG) from the circulation but leaves other plasma components unaltered.1,8
IVIg administration has widespread effects on a patient’s immune system by suppressing antibody production, inhibiting complement deposition and neutralising inflammatory cytokines and antibodies.1,3 The main difference between IVIg and SCIg is the lower absorption rate and initial bioavailability for SCIg compared to IVIg, as subcutaneously administered Ig first reaches the lymphatic system before entering the blood circulation.14
A number of new therapies for patients with MG are available or under clinical study, aiming to weaken the autoimmune response.1 These include:1
- Selective depletion of MG-specific autoantibodies during IgG apheresis
- Biologics that:
- Inhibit key complement components
- Inhibit receptors expressed by various immune cells
- Degrade pathogenic immunoglobulins involved in the pathophysiology of MG
- Autologous haematopoietic stem cell transplants in patients with severe or life-threatening MG
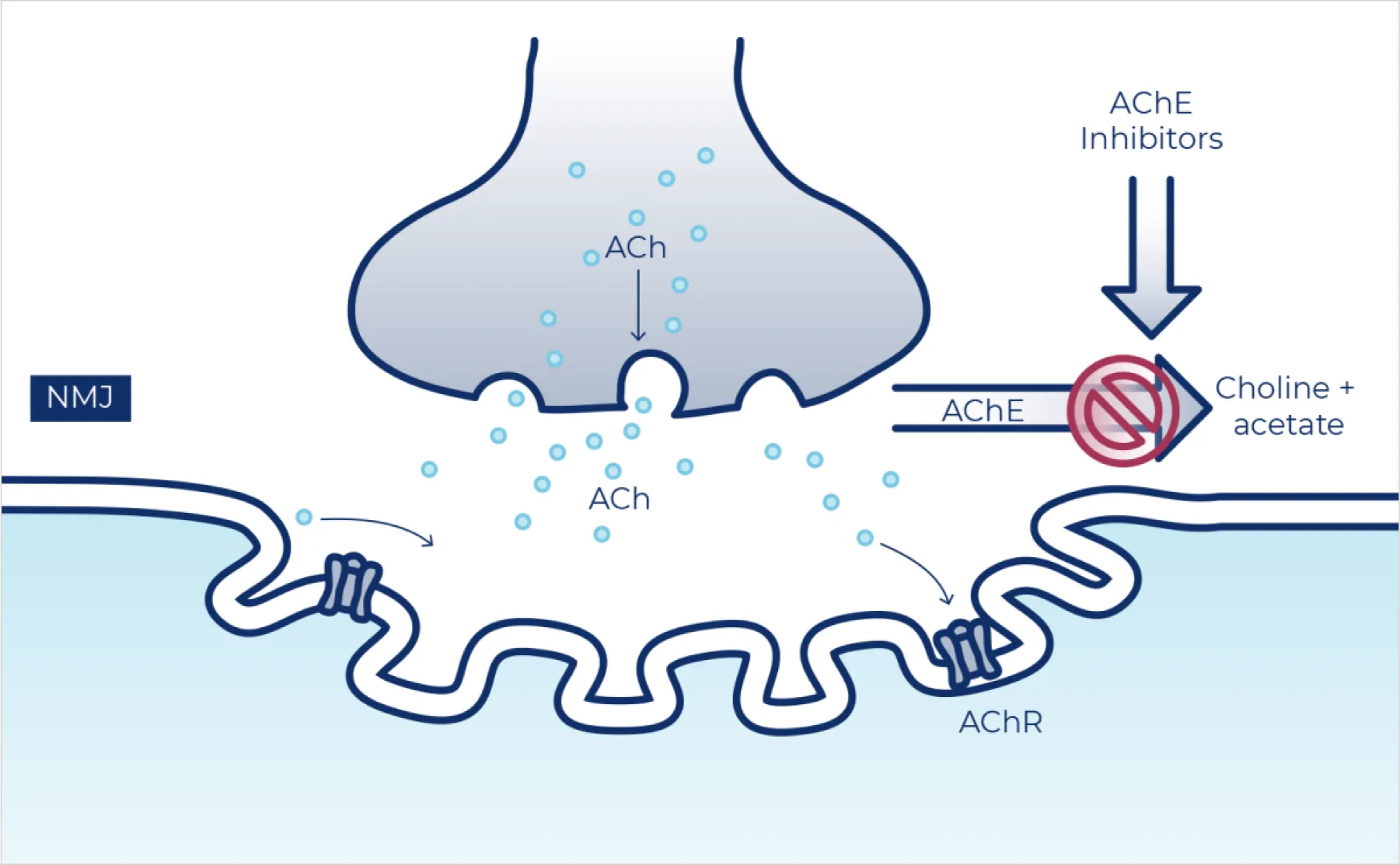
A smaller number of investigational therapies are aimed at strengthening the mechanisms involved at neuromuscular synapses.1
Another treatment goal of therapies that have been recently developed or are currently under development is to reduce adverse events related to conventional treatments, such as long-term or high-dose corticosteroid treatment.10,12,16