
Capire la NF1
Sebbene la NF1 sia una condizione genetica, solo la metà dei pazienti con il gene della variante la eredita da un genitore, mentre l'altra metà la sviluppa in modo casuale.1 Una persona con la variante ha il 50% di possibilità di trasmettere la variante ai propri figli.1
- Sei o più macchie "caffellatte" - macchie scolorite e colorate di caffè sulla pelle
- Lentiggini nell'area dell’ascella o dell'inguine
- Due o più neurofibromi di qualsiasi tipo o un neurofibroma plessiforme (PN) - tumori benigni che proliferano a partire dalle cellule che rivestono i nervi
- Glioma delle vie ottiche - un tumore cerebrale a lenta crescita intorno al nervo ottico
- 2 o più noduli di Lisch (macchie nell’iride), o 2 o più anomalie coroideali (macchie nella coroidea dell’occhio)2
- Una lesione ossea distintiva, un cambiamento anomalo di una o più ossa
- Il riscontro di una mutazione a carico del gene NF1 responsabile della malattia, presente nel 50% del tessuto apparentemente normale, come i globuli bianchi
1. Macchie "caffellatte"
Espandi per saperne di più
Riduci
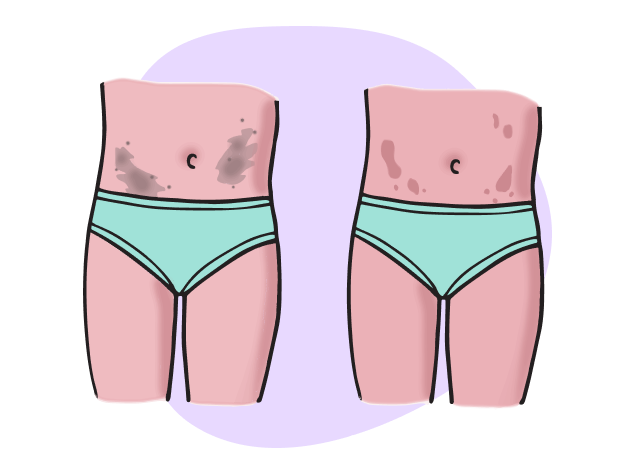
Le macchie caffè-au-lait, macchie scolorite e colorate di caffè sulla pelle, sono presenti nel 99% delle persone con NF1 e compaiono per la prima volta dalla nascita fino a 12 anni di età.4
2. Lentiggini
Espandi per saperne di più
Riduci
Nell’85% dei soggetti affetti da NF1 sono presenti lentiggini nelle ascelle o intorno all’inguine, che può manifestarsi per la prima volta dai 3 anni di età fino all’adolescenza.4

3. Neurofibromi
Espandi per saperne di più
Riduci
I neurofibromi sono tumori benigni che crescono lungo la lunghezza dei nervi.4 Possono essere superficiali (cutanei) o rimanere appena sotto la superficie (sottocutanei).4 I neurofibromi sono presenti nel 99% delle persone con NF1 e compaiono dopo i 7 anni di età, tipicamente nella tarda adolescenza.4
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono apparire dalla nascita fino a 18 anni di età.4

4. Glioma delle vie ottiche
Espandi per saperne di più
Riduci
I gliomi della via ottica sono tumori del nervo ottico che possono essere identificati mediante risonanza magnetica e sono stati osservati nel 15% delle persone con NF1.4,5 Compaiono per la prima volta dalla nascita fino a 7 anni di età.4

5. Anomalie dell’occhio
Espandi per saperne di più
Riduci
I segni di NF1 nell’occhio comprendono i noduli di lisch, ossia noduli che crescono nell’iride (la parte colorata dell’occhio) e sono presenti nel 90-95% delle persone con NF1.1,4 Tendono a comparire dopo 3 anni di età.4
Le anomalie coroideali sono macchie luminose e irregolari e possono essere rilevate anche mediante esami di imaging dell'occhio.2

6. Lesione ossea distintiva
Espandi per saperne di più
Riduci
Le lesioni ossee distintive comprendono difetti allo scheletro, per esempio, scoliosi (spina dorsale curva) e displasia delle ali sfenoidi (un osso che forma parte del cranio intorno agli occhi).1,4
La displasia dell'osso sfenoide è caratterizzata da un osso sfinoide di dimensioni ridotte o assente ed è presente in meno dell'1% delle persone con NF1.4,6 Se presente, si trova alla nascita.4
Altre lesioni distintive includono curvatura della tibia e fratture delle ossa lunghe che non guariscono.3

7. Mutazione del gene NF1
Espandi per saperne di più
Riduci
È possibile eseguire test genetici per identificare una variante del gene NF1 che causa la malattia.5 Questa variante deve essere presente nel 50% del tessuto apparentemente normale (ad es. globuli bianchi) per essere considerata indicativa di NF1.1

Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono causare una serie di sintomi, tra cui dolore, debolezza muscolare e deformità fisica.2,7 Puoi saperne di più su NF1 e PN nella pagina Vivere con la NF1.
Vivere con la NF1
Il PN può crescere lungo qualsiasi nervo e, a seconda del punto in cui cresce, può interessare diverse parti del corpo.7 Il PN è di solito benigno, ma in rari casi può diventare maligno.5
Nei bambini con NF1 non è sempre facile notare il PN. Talvolta si trovano in profondità all'interno del corpo, il che significa che non possono essere visti o sentiti.2 A volte possono essere riconosciuti da scolorimento della pelle o da un rigonfiamento sotto la pelle.2
Il PN può crescere rapidamente e può causare altri sintomi, tra cui:4,7,8
- Dolore
- Debolezza muscolare
- Compromissione visiva se il PN è vicino all’occhio
- Perdita del controllo della vescica
- Problemi intestinali, inclusa stipsi grave
- Problemi alle vie aeree, compresa la necessità di supporto meccanico
- Deformità fisica

Circa il 30-50% delle persone con NF1 sviluppa un PN.4 Il rilevamento precoce del PN semplifica la gestione e il trattamento della condizione.5 Se noti uno qualsiasi dei sintomi di cui sopra, rivolgiti al medico
Scopri come viene trattata NF1
Tamura R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci 2021(11);22:5850.
Bergqvist C et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis 2020;15(1):37.
Legius E et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med 2021;23(8):1506–1513.
Ferner RE et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44(2):81–88.
Williams VC et al. Neurofibromatosis type 1 revisited. Pediatrics 2009;123(1):124–133.
Radiopaedia.org. Sphenoid wing dysplasia. Available at: https://radiopaedia.org/articles/sphenoid-wing-dysplasia?lang=us Last accessed: September 2023.
Gross AM et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol 2018;20(12):1643–1651.
Copley-Merriman C et al. Natural history and disease burden of neurofibromatosis type 1 with plexiform neurofibromas: a systematic literature review. Adolesc Health Med Ther 2021;19:55–66.
Annulla
Continua
Le informazioni contenute in questo sito web sono fornite da Alexion, AstraZeneca Rare Disease come fonte di formazione generale a supporto dei pazienti e di chi se ne prende cura, esclusivamente nell'Unione Europea. Non sono destinate a scopi di autodiagnosi né a sostituire il parere del medico o di un operatore sanitario. Si prega di consultare il proprio medico o professionista sanitario per ulteriori informazioni o se si hanno domande sulla propria condizione medica. Tutte le fotografie utilizzate in questo sito web sono solo a scopo illustrativo..
Le macchie caffè-au-lait, macchie scolorite e colorate di caffè sulla pelle, sono presenti nel 99% delle persone con NF1 e compaiono per la prima volta dalla nascita fino a 12 anni di età.4
2. Lentiggini
Espandi per saperne di più
Riduci
Nell’85% dei soggetti affetti da NF1 sono presenti lentiggini nelle ascelle o intorno all’inguine, che può manifestarsi per la prima volta dai 3 anni di età fino all’adolescenza.4
3. Neurofibromi
Espandi per saperne di più
Riduci
I neurofibromi sono tumori benigni che crescono lungo la lunghezza dei nervi.4 Possono essere superficiali (cutanei) o rimanere appena sotto la superficie (sottocutanei).4 I neurofibromi sono presenti nel 99% delle persone con NF1 e compaiono dopo i 7 anni di età, tipicamente nella tarda adolescenza.4
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono apparire dalla nascita fino a 18 anni di età.4
4. Glioma delle vie ottiche
Espandi per saperne di più
Riduci
I gliomi della via ottica sono tumori del nervo ottico che possono essere identificati mediante risonanza magnetica e sono stati osservati nel 15% delle persone con NF1.4,5 Compaiono per la prima volta dalla nascita fino a 7 anni di età.4
5. Anomalie dell’occhio
Espandi per saperne di più
Riduci
I segni di NF1 nell’occhio comprendono i noduli di lisch, ossia noduli che crescono nell’iride (la parte colorata dell’occhio) e sono presenti nel 90-95% delle persone con NF1.1,4 Tendono a comparire dopo 3 anni di età.4
Le anomalie coroideali sono macchie luminose e irregolari e possono essere rilevate anche mediante esami di imaging dell'occhio.2
6. Lesione ossea distintiva
Espandi per saperne di più
Riduci
Le lesioni ossee distintive comprendono difetti allo scheletro, per esempio, scoliosi (spina dorsale curva) e displasia delle ali sfenoidi (un osso che forma parte del cranio intorno agli occhi).1,4
La displasia dell'osso sfenoide è caratterizzata da un osso sfinoide di dimensioni ridotte o assente ed è presente in meno dell'1% delle persone con NF1.4,6 Se presente, si trova alla nascita.4
Altre lesioni distintive includono curvatura della tibia e fratture delle ossa lunghe che non guariscono.3
7. Mutazione del gene NF1
Espandi per saperne di più
Riduci
È possibile eseguire test genetici per identificare una variante del gene NF1 che causa la malattia.5 Questa variante deve essere presente nel 50% del tessuto apparentemente normale (ad es. globuli bianchi) per essere considerata indicativa di NF1.1
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono causare una serie di sintomi, tra cui dolore, debolezza muscolare e deformità fisica.2,7 Puoi saperne di più su NF1 e PN nella pagina Vivere con la NF1.
Vivere con la NF1
Il PN può crescere lungo qualsiasi nervo e, a seconda del punto in cui cresce, può interessare diverse parti del corpo.7 Il PN è di solito benigno, ma in rari casi può diventare maligno.5
Nei bambini con NF1 non è sempre facile notare il PN. Talvolta si trovano in profondità all'interno del corpo, il che significa che non possono essere visti o sentiti.2 A volte possono essere riconosciuti da scolorimento della pelle o da un rigonfiamento sotto la pelle.2
Il PN può crescere rapidamente e può causare altri sintomi, tra cui:4,7,8
- Dolore
- Debolezza muscolare
- Compromissione visiva se il PN è vicino all’occhio
- Perdita del controllo della vescica
- Problemi intestinali, inclusa stipsi grave
- Problemi alle vie aeree, compresa la necessità di supporto meccanico
- Deformità fisica
Circa il 30-50% delle persone con NF1 sviluppa un PN.4 Il rilevamento precoce del PN semplifica la gestione e il trattamento della condizione.5 Se noti uno qualsiasi dei sintomi di cui sopra, rivolgiti al medico
Scopri come viene trattata NF1
Tamura R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci 2021(11);22:5850.
Bergqvist C et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis 2020;15(1):37.
Legius E et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med 2021;23(8):1506–1513.
Ferner RE et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44(2):81–88.
Williams VC et al. Neurofibromatosis type 1 revisited. Pediatrics 2009;123(1):124–133.
Radiopaedia.org. Sphenoid wing dysplasia. Available at: https://radiopaedia.org/articles/sphenoid-wing-dysplasia?lang=us Last accessed: September 2023.
Gross AM et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol 2018;20(12):1643–1651.
Copley-Merriman C et al. Natural history and disease burden of neurofibromatosis type 1 with plexiform neurofibromas: a systematic literature review. Adolesc Health Med Ther 2021;19:55–66.
Annulla
Continua
Le informazioni contenute in questo sito web sono fornite da Alexion, AstraZeneca Rare Disease come fonte di formazione generale a supporto dei pazienti e di chi se ne prende cura, esclusivamente nell'Unione Europea. Non sono destinate a scopi di autodiagnosi né a sostituire il parere del medico o di un operatore sanitario. Si prega di consultare il proprio medico o professionista sanitario per ulteriori informazioni o se si hanno domande sulla propria condizione medica. Tutte le fotografie utilizzate in questo sito web sono solo a scopo illustrativo..
Nell’85% dei soggetti affetti da NF1 sono presenti lentiggini nelle ascelle o intorno all’inguine, che può manifestarsi per la prima volta dai 3 anni di età fino all’adolescenza.4
3. Neurofibromi
Espandi per saperne di più
Riduci
I neurofibromi sono tumori benigni che crescono lungo la lunghezza dei nervi.4 Possono essere superficiali (cutanei) o rimanere appena sotto la superficie (sottocutanei).4 I neurofibromi sono presenti nel 99% delle persone con NF1 e compaiono dopo i 7 anni di età, tipicamente nella tarda adolescenza.4
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono apparire dalla nascita fino a 18 anni di età.4
4. Glioma delle vie ottiche
Espandi per saperne di più
Riduci
I gliomi della via ottica sono tumori del nervo ottico che possono essere identificati mediante risonanza magnetica e sono stati osservati nel 15% delle persone con NF1.4,5 Compaiono per la prima volta dalla nascita fino a 7 anni di età.4
5. Anomalie dell’occhio
Espandi per saperne di più
Riduci
I segni di NF1 nell’occhio comprendono i noduli di lisch, ossia noduli che crescono nell’iride (la parte colorata dell’occhio) e sono presenti nel 90-95% delle persone con NF1.1,4 Tendono a comparire dopo 3 anni di età.4
Le anomalie coroideali sono macchie luminose e irregolari e possono essere rilevate anche mediante esami di imaging dell'occhio.2
6. Lesione ossea distintiva
Espandi per saperne di più
Riduci
Le lesioni ossee distintive comprendono difetti allo scheletro, per esempio, scoliosi (spina dorsale curva) e displasia delle ali sfenoidi (un osso che forma parte del cranio intorno agli occhi).1,4
La displasia dell'osso sfenoide è caratterizzata da un osso sfinoide di dimensioni ridotte o assente ed è presente in meno dell'1% delle persone con NF1.4,6 Se presente, si trova alla nascita.4
Altre lesioni distintive includono curvatura della tibia e fratture delle ossa lunghe che non guariscono.3
7. Mutazione del gene NF1
Espandi per saperne di più
Riduci
È possibile eseguire test genetici per identificare una variante del gene NF1 che causa la malattia.5 Questa variante deve essere presente nel 50% del tessuto apparentemente normale (ad es. globuli bianchi) per essere considerata indicativa di NF1.1
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono causare una serie di sintomi, tra cui dolore, debolezza muscolare e deformità fisica.2,7 Puoi saperne di più su NF1 e PN nella pagina Vivere con la NF1.
Vivere con la NF1
Il PN può crescere lungo qualsiasi nervo e, a seconda del punto in cui cresce, può interessare diverse parti del corpo.7 Il PN è di solito benigno, ma in rari casi può diventare maligno.5
Nei bambini con NF1 non è sempre facile notare il PN. Talvolta si trovano in profondità all'interno del corpo, il che significa che non possono essere visti o sentiti.2 A volte possono essere riconosciuti da scolorimento della pelle o da un rigonfiamento sotto la pelle.2
Il PN può crescere rapidamente e può causare altri sintomi, tra cui:4,7,8
- Dolore
- Debolezza muscolare
- Compromissione visiva se il PN è vicino all’occhio
- Perdita del controllo della vescica
- Problemi intestinali, inclusa stipsi grave
- Problemi alle vie aeree, compresa la necessità di supporto meccanico
- Deformità fisica
Circa il 30-50% delle persone con NF1 sviluppa un PN.4 Il rilevamento precoce del PN semplifica la gestione e il trattamento della condizione.5 Se noti uno qualsiasi dei sintomi di cui sopra, rivolgiti al medico
Scopri come viene trattata NF1
Tamura R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci 2021(11);22:5850.
Bergqvist C et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis 2020;15(1):37.
Legius E et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med 2021;23(8):1506–1513.
Ferner RE et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44(2):81–88.
Williams VC et al. Neurofibromatosis type 1 revisited. Pediatrics 2009;123(1):124–133.
Radiopaedia.org. Sphenoid wing dysplasia. Available at: https://radiopaedia.org/articles/sphenoid-wing-dysplasia?lang=us Last accessed: September 2023.
Gross AM et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol 2018;20(12):1643–1651.
Copley-Merriman C et al. Natural history and disease burden of neurofibromatosis type 1 with plexiform neurofibromas: a systematic literature review. Adolesc Health Med Ther 2021;19:55–66.
Annulla
Continua
Le informazioni contenute in questo sito web sono fornite da Alexion, AstraZeneca Rare Disease come fonte di formazione generale a supporto dei pazienti e di chi se ne prende cura, esclusivamente nell'Unione Europea. Non sono destinate a scopi di autodiagnosi né a sostituire il parere del medico o di un operatore sanitario. Si prega di consultare il proprio medico o professionista sanitario per ulteriori informazioni o se si hanno domande sulla propria condizione medica. Tutte le fotografie utilizzate in questo sito web sono solo a scopo illustrativo..
I neurofibromi sono tumori benigni che crescono lungo la lunghezza dei nervi.4 Possono essere superficiali (cutanei) o rimanere appena sotto la superficie (sottocutanei).4 I neurofibromi sono presenti nel 99% delle persone con NF1 e compaiono dopo i 7 anni di età, tipicamente nella tarda adolescenza.4
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono apparire dalla nascita fino a 18 anni di età.4
4. Glioma delle vie ottiche
Espandi per saperne di più
Riduci
I gliomi della via ottica sono tumori del nervo ottico che possono essere identificati mediante risonanza magnetica e sono stati osservati nel 15% delle persone con NF1.4,5 Compaiono per la prima volta dalla nascita fino a 7 anni di età.4
5. Anomalie dell’occhio
Espandi per saperne di più
Riduci
I segni di NF1 nell’occhio comprendono i noduli di lisch, ossia noduli che crescono nell’iride (la parte colorata dell’occhio) e sono presenti nel 90-95% delle persone con NF1.1,4 Tendono a comparire dopo 3 anni di età.4
Le anomalie coroideali sono macchie luminose e irregolari e possono essere rilevate anche mediante esami di imaging dell'occhio.2
6. Lesione ossea distintiva
Espandi per saperne di più
Riduci
Le lesioni ossee distintive comprendono difetti allo scheletro, per esempio, scoliosi (spina dorsale curva) e displasia delle ali sfenoidi (un osso che forma parte del cranio intorno agli occhi).1,4
La displasia dell'osso sfenoide è caratterizzata da un osso sfinoide di dimensioni ridotte o assente ed è presente in meno dell'1% delle persone con NF1.4,6 Se presente, si trova alla nascita.4
Altre lesioni distintive includono curvatura della tibia e fratture delle ossa lunghe che non guariscono.3
7. Mutazione del gene NF1
Espandi per saperne di più
Riduci
È possibile eseguire test genetici per identificare una variante del gene NF1 che causa la malattia.5 Questa variante deve essere presente nel 50% del tessuto apparentemente normale (ad es. globuli bianchi) per essere considerata indicativa di NF1.1
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono causare una serie di sintomi, tra cui dolore, debolezza muscolare e deformità fisica.2,7 Puoi saperne di più su NF1 e PN nella pagina Vivere con la NF1.
Vivere con la NF1
Il PN può crescere lungo qualsiasi nervo e, a seconda del punto in cui cresce, può interessare diverse parti del corpo.7 Il PN è di solito benigno, ma in rari casi può diventare maligno.5
Nei bambini con NF1 non è sempre facile notare il PN. Talvolta si trovano in profondità all'interno del corpo, il che significa che non possono essere visti o sentiti.2 A volte possono essere riconosciuti da scolorimento della pelle o da un rigonfiamento sotto la pelle.2
Il PN può crescere rapidamente e può causare altri sintomi, tra cui:4,7,8
- Dolore
- Debolezza muscolare
- Compromissione visiva se il PN è vicino all’occhio
- Perdita del controllo della vescica
- Problemi intestinali, inclusa stipsi grave
- Problemi alle vie aeree, compresa la necessità di supporto meccanico
- Deformità fisica
Circa il 30-50% delle persone con NF1 sviluppa un PN.4 Il rilevamento precoce del PN semplifica la gestione e il trattamento della condizione.5 Se noti uno qualsiasi dei sintomi di cui sopra, rivolgiti al medico
Scopri come viene trattata NF1
Tamura R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci 2021(11);22:5850.
Bergqvist C et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis 2020;15(1):37.
Legius E et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med 2021;23(8):1506–1513.
Ferner RE et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44(2):81–88.
Williams VC et al. Neurofibromatosis type 1 revisited. Pediatrics 2009;123(1):124–133.
Radiopaedia.org. Sphenoid wing dysplasia. Available at: https://radiopaedia.org/articles/sphenoid-wing-dysplasia?lang=us Last accessed: September 2023.
Gross AM et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol 2018;20(12):1643–1651.
Copley-Merriman C et al. Natural history and disease burden of neurofibromatosis type 1 with plexiform neurofibromas: a systematic literature review. Adolesc Health Med Ther 2021;19:55–66.
Annulla
Continua
Le informazioni contenute in questo sito web sono fornite da Alexion, AstraZeneca Rare Disease come fonte di formazione generale a supporto dei pazienti e di chi se ne prende cura, esclusivamente nell'Unione Europea. Non sono destinate a scopi di autodiagnosi né a sostituire il parere del medico o di un operatore sanitario. Si prega di consultare il proprio medico o professionista sanitario per ulteriori informazioni o se si hanno domande sulla propria condizione medica. Tutte le fotografie utilizzate in questo sito web sono solo a scopo illustrativo..
I gliomi della via ottica sono tumori del nervo ottico che possono essere identificati mediante risonanza magnetica e sono stati osservati nel 15% delle persone con NF1.4,5 Compaiono per la prima volta dalla nascita fino a 7 anni di età.4
5. Anomalie dell’occhio
Espandi per saperne di più
Riduci
I segni di NF1 nell’occhio comprendono i noduli di lisch, ossia noduli che crescono nell’iride (la parte colorata dell’occhio) e sono presenti nel 90-95% delle persone con NF1.1,4 Tendono a comparire dopo 3 anni di età.4
Le anomalie coroideali sono macchie luminose e irregolari e possono essere rilevate anche mediante esami di imaging dell'occhio.2
6. Lesione ossea distintiva
Espandi per saperne di più
Riduci
Le lesioni ossee distintive comprendono difetti allo scheletro, per esempio, scoliosi (spina dorsale curva) e displasia delle ali sfenoidi (un osso che forma parte del cranio intorno agli occhi).1,4
La displasia dell'osso sfenoide è caratterizzata da un osso sfinoide di dimensioni ridotte o assente ed è presente in meno dell'1% delle persone con NF1.4,6 Se presente, si trova alla nascita.4
Altre lesioni distintive includono curvatura della tibia e fratture delle ossa lunghe che non guariscono.3
7. Mutazione del gene NF1
Espandi per saperne di più
Riduci
È possibile eseguire test genetici per identificare una variante del gene NF1 che causa la malattia.5 Questa variante deve essere presente nel 50% del tessuto apparentemente normale (ad es. globuli bianchi) per essere considerata indicativa di NF1.1
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono causare una serie di sintomi, tra cui dolore, debolezza muscolare e deformità fisica.2,7 Puoi saperne di più su NF1 e PN nella pagina Vivere con la NF1.
Vivere con la NF1
Il PN può crescere lungo qualsiasi nervo e, a seconda del punto in cui cresce, può interessare diverse parti del corpo.7 Il PN è di solito benigno, ma in rari casi può diventare maligno.5
Nei bambini con NF1 non è sempre facile notare il PN. Talvolta si trovano in profondità all'interno del corpo, il che significa che non possono essere visti o sentiti.2 A volte possono essere riconosciuti da scolorimento della pelle o da un rigonfiamento sotto la pelle.2
Il PN può crescere rapidamente e può causare altri sintomi, tra cui:4,7,8
- Dolore
- Debolezza muscolare
- Compromissione visiva se il PN è vicino all’occhio
- Perdita del controllo della vescica
- Problemi intestinali, inclusa stipsi grave
- Problemi alle vie aeree, compresa la necessità di supporto meccanico
- Deformità fisica
Circa il 30-50% delle persone con NF1 sviluppa un PN.4 Il rilevamento precoce del PN semplifica la gestione e il trattamento della condizione.5 Se noti uno qualsiasi dei sintomi di cui sopra, rivolgiti al medico
Scopri come viene trattata NF1
Tamura R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci 2021(11);22:5850.
Bergqvist C et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis 2020;15(1):37.
Legius E et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med 2021;23(8):1506–1513.
Ferner RE et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44(2):81–88.
Williams VC et al. Neurofibromatosis type 1 revisited. Pediatrics 2009;123(1):124–133.
Radiopaedia.org. Sphenoid wing dysplasia. Available at: https://radiopaedia.org/articles/sphenoid-wing-dysplasia?lang=us Last accessed: September 2023.
Gross AM et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol 2018;20(12):1643–1651.
Copley-Merriman C et al. Natural history and disease burden of neurofibromatosis type 1 with plexiform neurofibromas: a systematic literature review. Adolesc Health Med Ther 2021;19:55–66.
Annulla
Continua
Le informazioni contenute in questo sito web sono fornite da Alexion, AstraZeneca Rare Disease come fonte di formazione generale a supporto dei pazienti e di chi se ne prende cura, esclusivamente nell'Unione Europea. Non sono destinate a scopi di autodiagnosi né a sostituire il parere del medico o di un operatore sanitario. Si prega di consultare il proprio medico o professionista sanitario per ulteriori informazioni o se si hanno domande sulla propria condizione medica. Tutte le fotografie utilizzate in questo sito web sono solo a scopo illustrativo..
I segni di NF1 nell’occhio comprendono i noduli di lisch, ossia noduli che crescono nell’iride (la parte colorata dell’occhio) e sono presenti nel 90-95% delle persone con NF1.1,4 Tendono a comparire dopo 3 anni di età.4
Le anomalie coroideali sono macchie luminose e irregolari e possono essere rilevate anche mediante esami di imaging dell'occhio.2
6. Lesione ossea distintiva
Espandi per saperne di più
Riduci
Le lesioni ossee distintive comprendono difetti allo scheletro, per esempio, scoliosi (spina dorsale curva) e displasia delle ali sfenoidi (un osso che forma parte del cranio intorno agli occhi).1,4
La displasia dell'osso sfenoide è caratterizzata da un osso sfinoide di dimensioni ridotte o assente ed è presente in meno dell'1% delle persone con NF1.4,6 Se presente, si trova alla nascita.4
Altre lesioni distintive includono curvatura della tibia e fratture delle ossa lunghe che non guariscono.3
7. Mutazione del gene NF1
Espandi per saperne di più
Riduci
È possibile eseguire test genetici per identificare una variante del gene NF1 che causa la malattia.5 Questa variante deve essere presente nel 50% del tessuto apparentemente normale (ad es. globuli bianchi) per essere considerata indicativa di NF1.1
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono causare una serie di sintomi, tra cui dolore, debolezza muscolare e deformità fisica.2,7 Puoi saperne di più su NF1 e PN nella pagina Vivere con la NF1.
Vivere con la NF1
Il PN può crescere lungo qualsiasi nervo e, a seconda del punto in cui cresce, può interessare diverse parti del corpo.7 Il PN è di solito benigno, ma in rari casi può diventare maligno.5
Nei bambini con NF1 non è sempre facile notare il PN. Talvolta si trovano in profondità all'interno del corpo, il che significa che non possono essere visti o sentiti.2 A volte possono essere riconosciuti da scolorimento della pelle o da un rigonfiamento sotto la pelle.2
Il PN può crescere rapidamente e può causare altri sintomi, tra cui:4,7,8
- Dolore
- Debolezza muscolare
- Compromissione visiva se il PN è vicino all’occhio
- Perdita del controllo della vescica
- Problemi intestinali, inclusa stipsi grave
- Problemi alle vie aeree, compresa la necessità di supporto meccanico
- Deformità fisica
Circa il 30-50% delle persone con NF1 sviluppa un PN.4 Il rilevamento precoce del PN semplifica la gestione e il trattamento della condizione.5 Se noti uno qualsiasi dei sintomi di cui sopra, rivolgiti al medico
Scopri come viene trattata NF1
Tamura R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci 2021(11);22:5850.
Bergqvist C et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis 2020;15(1):37.
Legius E et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med 2021;23(8):1506–1513.
Ferner RE et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44(2):81–88.
Williams VC et al. Neurofibromatosis type 1 revisited. Pediatrics 2009;123(1):124–133.
Radiopaedia.org. Sphenoid wing dysplasia. Available at: https://radiopaedia.org/articles/sphenoid-wing-dysplasia?lang=us Last accessed: September 2023.
Gross AM et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol 2018;20(12):1643–1651.
Copley-Merriman C et al. Natural history and disease burden of neurofibromatosis type 1 with plexiform neurofibromas: a systematic literature review. Adolesc Health Med Ther 2021;19:55–66.
Annulla
Continua
Le informazioni contenute in questo sito web sono fornite da Alexion, AstraZeneca Rare Disease come fonte di formazione generale a supporto dei pazienti e di chi se ne prende cura, esclusivamente nell'Unione Europea. Non sono destinate a scopi di autodiagnosi né a sostituire il parere del medico o di un operatore sanitario. Si prega di consultare il proprio medico o professionista sanitario per ulteriori informazioni o se si hanno domande sulla propria condizione medica. Tutte le fotografie utilizzate in questo sito web sono solo a scopo illustrativo..
Le lesioni ossee distintive comprendono difetti allo scheletro, per esempio, scoliosi (spina dorsale curva) e displasia delle ali sfenoidi (un osso che forma parte del cranio intorno agli occhi).1,4
La displasia dell'osso sfenoide è caratterizzata da un osso sfinoide di dimensioni ridotte o assente ed è presente in meno dell'1% delle persone con NF1.4,6 Se presente, si trova alla nascita.4
Altre lesioni distintive includono curvatura della tibia e fratture delle ossa lunghe che non guariscono.3
7. Mutazione del gene NF1
Espandi per saperne di più
Riduci
È possibile eseguire test genetici per identificare una variante del gene NF1 che causa la malattia.5 Questa variante deve essere presente nel 50% del tessuto apparentemente normale (ad es. globuli bianchi) per essere considerata indicativa di NF1.1
Il neurofibroma plessiforme (PN) è un tumore che cresce sui nervi. I PN sono presenti in circa il 30% delle persone affette da NF1.4,5 Possono causare una serie di sintomi, tra cui dolore, debolezza muscolare e deformità fisica.2,7 Puoi saperne di più su NF1 e PN nella pagina Vivere con la NF1.
Vivere con la NF1
Il PN può crescere lungo qualsiasi nervo e, a seconda del punto in cui cresce, può interessare diverse parti del corpo.7 Il PN è di solito benigno, ma in rari casi può diventare maligno.5
Nei bambini con NF1 non è sempre facile notare il PN. Talvolta si trovano in profondità all'interno del corpo, il che significa che non possono essere visti o sentiti.2 A volte possono essere riconosciuti da scolorimento della pelle o da un rigonfiamento sotto la pelle.2
Il PN può crescere rapidamente e può causare altri sintomi, tra cui:4,7,8
- Dolore
- Debolezza muscolare
- Compromissione visiva se il PN è vicino all’occhio
- Perdita del controllo della vescica
- Problemi intestinali, inclusa stipsi grave
- Problemi alle vie aeree, compresa la necessità di supporto meccanico
- Deformità fisica
Circa il 30-50% delle persone con NF1 sviluppa un PN.4 Il rilevamento precoce del PN semplifica la gestione e il trattamento della condizione.5 Se noti uno qualsiasi dei sintomi di cui sopra, rivolgiti al medico
Scopri come viene trattata NF1
Tamura R. Current understanding of neurofibromatosis type 1, 2, and schwannomatosis. Int J Mol Sci 2021(11);22:5850.
Bergqvist C et al. Neurofibromatosis 1 French national guidelines based on an extensive literature review since 1966. Orphanet J Rare Dis 2020;15(1):37.
Legius E et al. Revised diagnostic criteria for neurofibromatosis type 1 and Legius syndrome: An international consensus recommendation. Genet Med 2021;23(8):1506–1513.
Ferner RE et al. Guidelines for the diagnosis and management of individuals with neurofibromatosis 1. J Med Genet 2007;44(2):81–88.
Williams VC et al. Neurofibromatosis type 1 revisited. Pediatrics 2009;123(1):124–133.
Radiopaedia.org. Sphenoid wing dysplasia. Available at: https://radiopaedia.org/articles/sphenoid-wing-dysplasia?lang=us Last accessed: September 2023.
Gross AM et al. Association of plexiform neurofibroma volume changes and development of clinical morbidities in neurofibromatosis 1. Neuro Oncol 2018;20(12):1643–1651.
Copley-Merriman C et al. Natural history and disease burden of neurofibromatosis type 1 with plexiform neurofibromas: a systematic literature review. Adolesc Health Med Ther 2021;19:55–66.
Annulla
Continua
Le informazioni contenute in questo sito web sono fornite da Alexion, AstraZeneca Rare Disease come fonte di formazione generale a supporto dei pazienti e di chi se ne prende cura, esclusivamente nell'Unione Europea. Non sono destinate a scopi di autodiagnosi né a sostituire il parere del medico o di un operatore sanitario. Si prega di consultare il proprio medico o professionista sanitario per ulteriori informazioni o se si hanno domande sulla propria condizione medica. Tutte le fotografie utilizzate in questo sito web sono solo a scopo illustrativo..
È possibile eseguire test genetici per identificare una variante del gene NF1 che causa la malattia.5 Questa variante deve essere presente nel 50% del tessuto apparentemente normale (ad es. globuli bianchi) per essere considerata indicativa di NF1.1
Nei bambini con NF1 non è sempre facile notare il PN. Talvolta si trovano in profondità all'interno del corpo, il che significa che non possono essere visti o sentiti.2 A volte possono essere riconosciuti da scolorimento della pelle o da un rigonfiamento sotto la pelle.2
Il PN può crescere rapidamente e può causare altri sintomi, tra cui:4,7,8
- Dolore
- Debolezza muscolare
- Compromissione visiva se il PN è vicino all’occhio
- Perdita del controllo della vescica
- Problemi intestinali, inclusa stipsi grave
- Problemi alle vie aeree, compresa la necessità di supporto meccanico
- Deformità fisica