
Neurofibromatosis type 1 (NF1) is a relatively common genetic condition that is estimated to occur in 1 in 3000 live births worldwide and causes tumours to grow along nerves.1–3
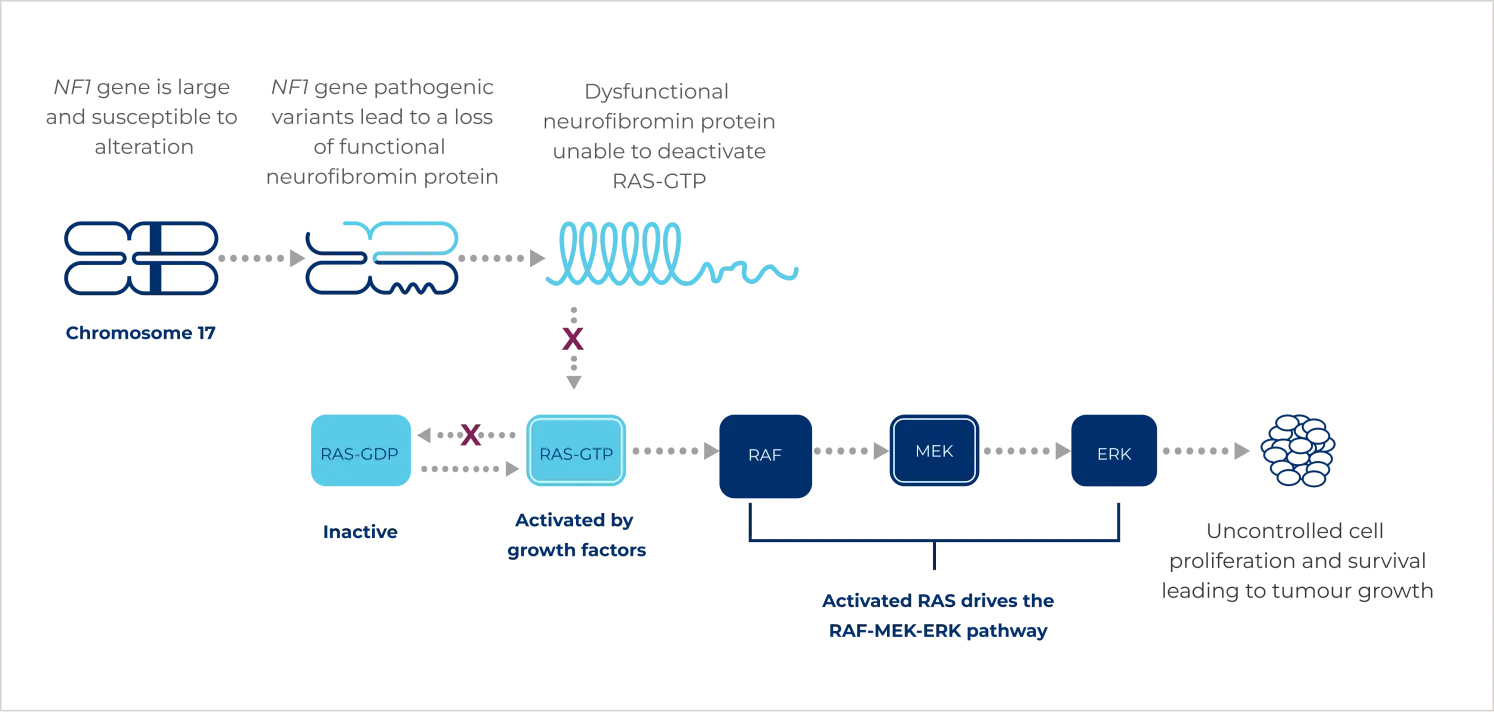
It is an autosomal dominant condition driven by pathogenic variants of the NF1 gene. Pathogenic variants can either be inherited or can occur spontaneously.2 These variants result in the production of dysfunctional or less active neurofibromin, the product of the NF1 gene.4 Neurofibromin is a tumour suppressor protein that regulates the constitutively active rat sarcoma viral oncogene homologue (RAS) protein by keeping it in an inactive form and preventing excessive stimulation of multiple proliferative cellular pathways. Pathogenic variants in the NF1 gene lead to constitutive activation of the RAS- rapidly accelerated fibrosarcoma (RAF)- mitogen-activated protein kinase kinase (MEK)- extracellular signal-regulated kinase (ERK) pathway and uncontrolled growth signalling.4
{kind=link}

Approximately 30–50% patients with NF1 have PN, a tumour of peripheral nerves arising from a bundle of fascicles or a larger nerve plexus.5 PN may remain asymptomatic or can invade surrounding muscle and bone, causing substantial pain and disfigurement.1
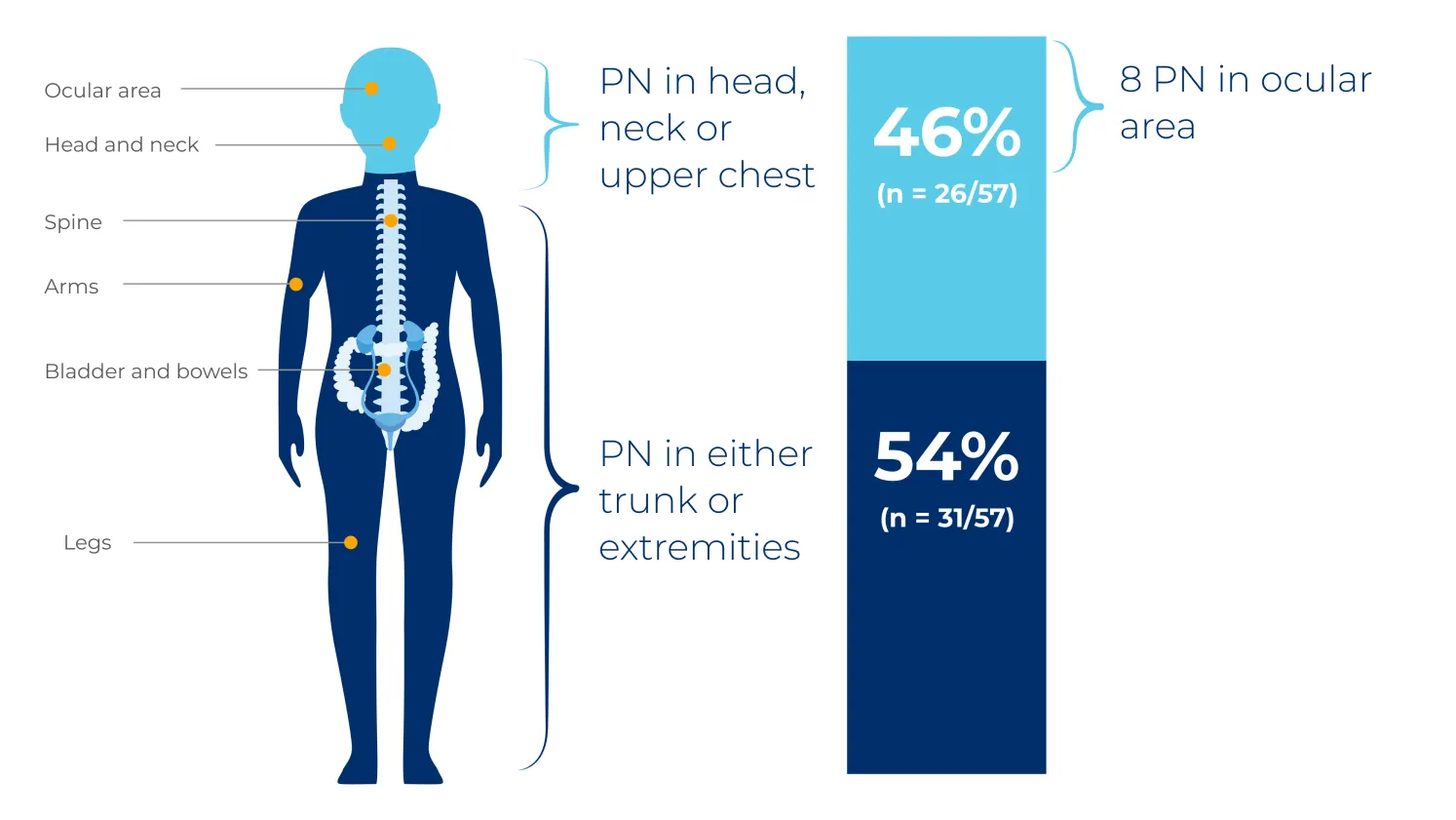
PN are considered congenital and may grow most rapidly in the first decade of life.5 They commonly occur in the head and neck region, trunk or extremities, but can affect nerves between the spinal root and the distal periphery.4,6
{kind=link}
The NCI natural history study was a US-based retrospective review of 41 patients with NF1, assessing PN volume and PN-related morbidities in 57 distinct PN over 7 years.6
